

ISO 13485: Integrating Regulatory Frameworks from Day One in Medical Device Development
Discover why early ISO 13485 integration is strategic for MedTech R&D. Ensure CE marking & FDA compliance for your minimally invasive surgical devices from day one.
Camille S. Quality manager Regulatory Compliance Engineer
2/17/20265 min read
The development of medical devices, particularly in the specialized field of minimally invasive surgery (MIS), requires a precise alignment between engineering innovation and clinical safety. For R&D engineers, startup founders, and clinicians, the transition from a functional prototype to a marketable and certified medical device involves navigating a complex regulatory landscape. Central to this journey is ISO 13485, the international standard for Medical Device Quality Management Systems (QMS). Establishing this framework from the earliest stages of development is a strategic decision that supports both technical excellence and commercial scalability.
Understanding the ISO 13485 Standard
ISO 13485 is a standalone QMS standard, derived from the broader ISO 9000 series but tailored specifically to the medical device industry. It provides a comprehensive framework for organizations to demonstrate their ability to provide medical devices and related services that consistently meet customer and applicable regulatory requirements. Unlike general quality standards, ISO 13485 emphasizes risk management, design controls, and production environments, elements that are critical in the development of surgical instrumentation.
The standard applies to all stages of the life cycle, including design and development, production, storage and distribution, installation, and servicing. For a startup or an R&D department, adopting ISO 13485 means implementing a systematic approach to documentation and process control. This ensures that every decision made during the development of a device is traceable, repeatable, and focused on patient safety.
Distinguishing ISO 13485 from the Medical Device Regulation (MDR)
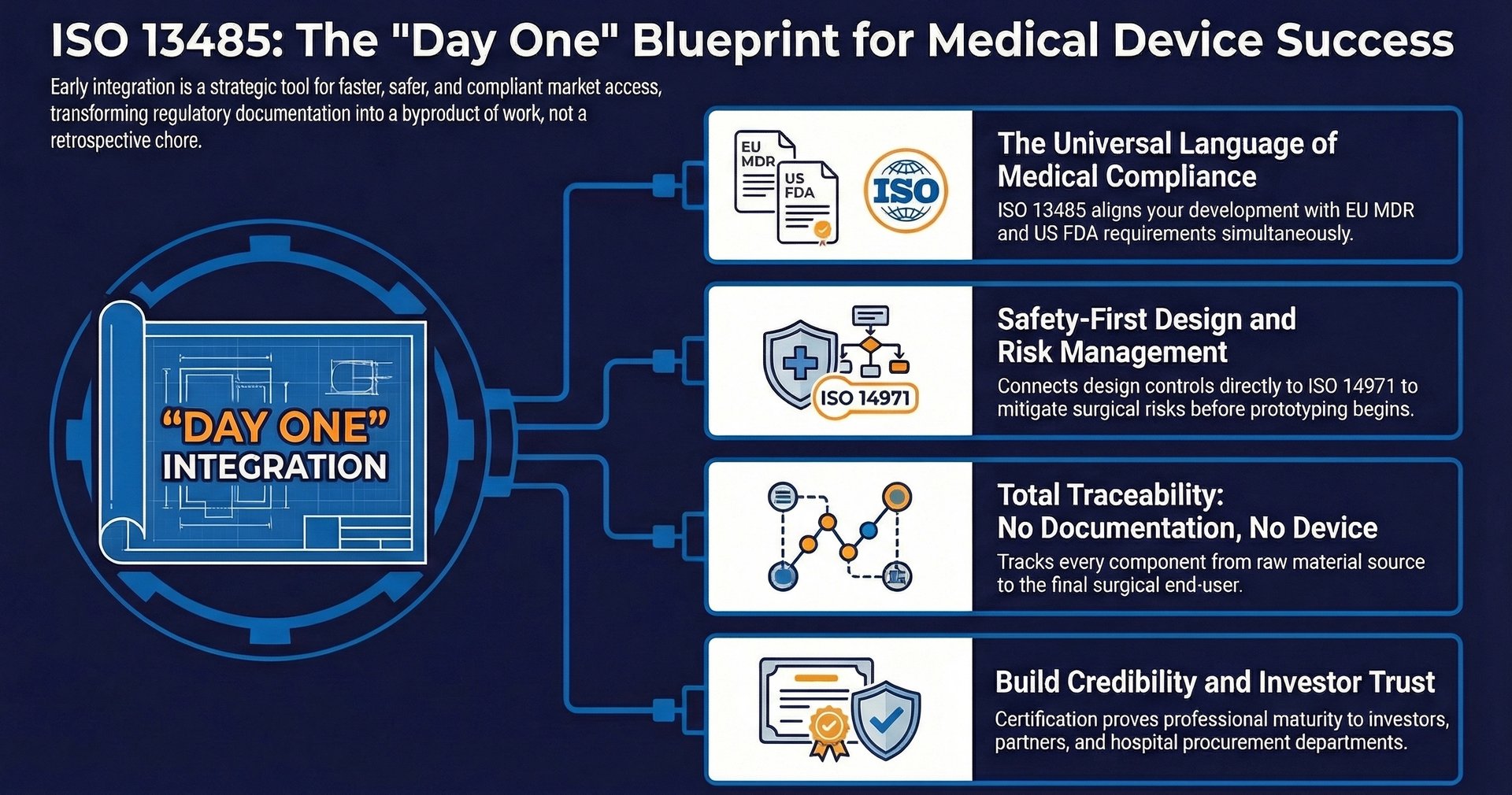
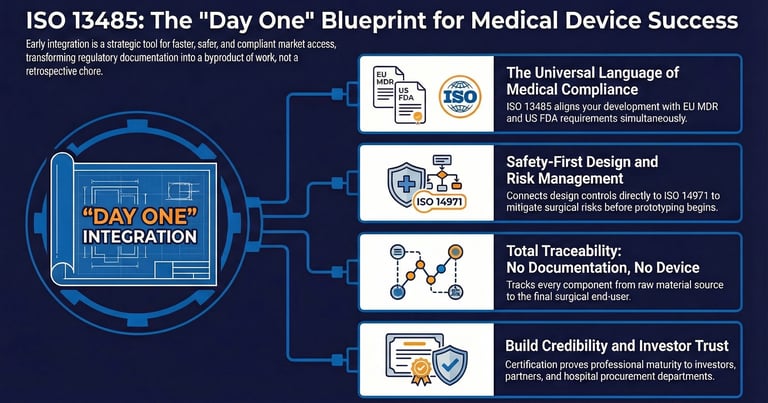
A common point of discussion in the European medical sector is the relationship between ISO 13485 and the European Medical Device Regulation (MDR 2017/745). While they are distinct, they function in a complementary manner. The MDR is a legal requirement: a regulation that must be followed to place a device on the European market. Since its harmonization with MDR 2017/745, compliance with ISO 13485:2016 enables a large part of the QMS requirements of the regulation to be met.
Choosing ISO 13485 as the foundation of your quality system is the most recognized path to compliance. While the MDR specifies what must be achieved to place a medical device on the market (such as clinical evaluation and post-market surveillance), ISO 13485 describes how to manage the internal processes to reach this goalISO13485 is the global standard for medical device, and many local regulations are based on or supplement its provisions such as the FDA’s Quality System Regulation (QSR) in the United States.
The Role of ISO 13485 in CE Marking and FDA Clearance
For any medical device to reach the clinical setting, it must undergo assessment by regulatory bodies: the Notified Bodies for CE marking in Europe or the FDA for 510(k) or PMA pathways in the US. ISO 13485 serves as a universal language between the manufacturer and these authorities.
In the European Union, compliance with ISO 13485 creates a presumption of conformity with the QMS requirements of the MDR. Similarly, the FDA has recently moved to align its 21 CFR Part 820 regulations more closely with ISO 13485 through the Quality System Regulation Amendment (QSRA).
By implementing ISO 13485 early, a company builds the "Technical Documentation" and "Design History File" (DHF) required for these submissions in a natural, incremental fashion. This alignment facilitates smoother audits and reduces the time required for regulatory review.
Practical Implementation: QMS, Standards, and Reporting Core pillars for fast and secure market access
In practical terms, implementing ISO 13485 from the start involves establishing several core pillars within the organization. These pillars provide the structure necessary for high-stakes projects like minimally invasive surgical tools. While they cover such areas as staff training and management commitments, here we highlight those that are relevant to successful development.
Design Controls and Risk Management
The standard requires a rigorous design and development process. This includes defined design inputs (what the device must do), design outputs (a design and its specifications), and verification and validation activities. Verification ensures the device is built correctly, while validation ensures the correct device was built for the user's needs. Notably, we can highlight the link to ISO 14971 on risk management, which provides for the identification and mitigation of risks from the start of the project by enabling the earliest possible identification of potential failure modes in a surgical procedure, as early as the design input stage.
Controlled Documentation and Traceability
ISO 13485 emphasizes that "if it is not documented, it did not happen." This involves maintaining a Device Master Record (DMR) which contains the instructions for manufacturing and testing the device.
There is also talk of maintaining the medical device file, which will make it possible to trace the entire life of the device, from its latest marketed improvement to the origin of the initial idea.
Traceability is not just a matter of design, but also of routine: where it is equally vital; for an MIS device, this means being able to trace every component back to its raw material source and forward to the end-user.
Corrective Actions and Preventive Actions (CAPA)
The QMS includes a feedback loop known as CAPA. This process allows the team to eliminate any non-conformity, to investigate there root causes, whether found during testing or via clinician feedback, and implement changes to prevent recurrence. This creates a culture of continuous improvement that enhances the reliability of the device.
The Advantages of Early-Stage Regulatory Integration
Integrating the regulatory framework from "Day One" is often viewed through the lens of efficiency. When a development team operates within an ISO 13485-compliant environment from the beginning, the documentation required for a regulatory submission is a byproduct of the work rather than a separate, retrospective task.
Early integration allows for the identification of applicable "Vertical Standards", specific norms for particular types of devices, such as ISO 60601 for electrical medical equipment or ISO 10555-1 for sterile and single-use catheters. Knowing these requirements during the initial sketching phase prevents design choices that might later be found non-compliant. This proactive approach ensures that the path from the laboratory to the operating room is predictable and characterized by steady progress.
Initial Steps for Establishing an ISO 13485 Framework
Starting the journey toward ISO 13485 compliance is a manageable process when broken down into logical phases. For many startups and R&D teams, the most effective path involves a blend of internal commitment and external expertise.
1. Gap Analysis: Evaluate existing processes against the requirements of the standard to identify what is already in place and what needs to be developed.
2. Define Scope: Clearly state the types of devices and activities the QMS will cover. For a firm focused on MIS, this would include design and potentially small-batch prototyping.
3. Establish Quality Policy and Objectives: Align the organization’s goals with commitment to quality and regulatory compliance.
4. Process Mapping: Document the core processes of the organization, from how requirements are captured from clinicians to how suppliers are selected and managed.
5. Training: Ensure that the engineering and clinical teams understand their roles within the QMS. Compliance is a collective responsibility.
Building Credibility through Technical Rigor
For founders and clinicians, the ultimate goal is to bring a solution to market that improves patient outcomes. An ISO 13485-certified framework is a powerful indicator of professional maturity. It demonstrates to investors, partners, and hospital procurement departments that the device has been developed under a controlled, high-quality regime. In the field of surgery, where precision is paramount, this technical rigor is the foundation of trust.
By treating the regulatory framework as a tool for better engineering rather than a bureaucratic hurdle, teams can focus on what they do best: innovating for the future of healthcare.


© 2026. All rights reserved.
Site map
Contract medical device development in cardiovascular and minimally invasive surgery.

