

How to choose a medical device engineering partner: the founder's checklist
How to choose the right engineering partner for your medical device? CDO or CDMO, real QMS or window dressing, in-house prototyping, regulatory approach, team continuity: eight concrete criteria, the right questions to ask and the red flags to watch out for, from those who see the same mistakes repeat themselves.
Yann Hoffbeck
3/20/202615 min read
Most founders who come to us have already made at least one expensive mistake with a previous engineering partner. Not because they were naive. Because choosing an engineering firm for a medical device is genuinely hard and the signals that look good on paper (certifications, client logos, slick website) don't tell you much about what actually matters.
This article is a checklist. Eight criteria. Each one comes from something we've seen go wrong in real projects. We're going to be direct about what those things are, what questions to ask, and what answers should make you nervous.
We'll also be honest about where we stand on each criterion ourselves because a checklist that only helps you evaluate other firms while conveniently ignoring us would be pretty useless.
Who this is for
Medtech founders, clinical entrepreneurs, and R&D project managers evaluating engineering partners for minimally invasive device development at any stage from concept to design freeze.
1. Are you talking to a CDO or a CDMO? and do you know the difference?
This distinction matters more than most people realize when they start looking for an engineering partner.
A CDMO: Contract Development and Manufacturing Organization, is fundamentally an industrial operator. Their business model runs on factory throughput. They have machines, production lines, and a workforce built around volume. The development side exists, but it's a means to an end: getting your device into their manufacturing process. Their commercial incentive will naturally pull solutions toward what they already know how to produce.
That's not a criticism. CDMOs are essential. But they come in at the right moment: when your design is frozen and you're scaling production. Before that, their involvement can distort technical choices in ways that are hard to see until it's too late.
A CDO: Contract Development Organization, has no factory to feed. No internal manufacturing strategy to protect. Their only interest is finding the right solution for your specific project, which sometimes means recommending a manufacturing process they don't run, a material they don't stock, or a CDMO that isn't them. That's what agnostic really means in this context.
The cleanest way to think about it: a CDO works for you. A CDMO works with you, but they also work for their own production capacity.
What to ask
– Do you have manufacturing capabilities? If yes, how do you ensure those capabilities don't influence design recommendations?
– Have you ever recommended a manufacturing partner outside your own organization? Why?
– At what point in the project do you typically introduce your manufacturing partners?
Red flag: "We handle everything and we won't tell you why."
Vertical integration can be a genuine strength: faster iteration, tighter quality control, and smoother handoffs. The real red flag is when a firm steers design decisions toward their own capabilities without being transparent about it. Always ask: what happens if the optimal design requires a process you don't run in-house? A trustworthy partner will give you an honest answer, and tell you when to look elsewhere.
Protomed is a CDO. We have no manufacturing operations. When a project reaches industrialization, we help our clients choose the right CDMO based on their device's requirements, not ours. With over 20 years of experience, we have built a large network of trusted industrial in North America Europe and Asia. We know who’s the best to manufacture what you need, and who, we think, you should avoid.
2. Is the ISO 13485 certification backed by a real quality team, or is it just a certificate on the wall?
ISO 13485 requires a Quality Management System. Getting certified means an external auditor confirmed that system exists and functions. That's real. It matters.
What it doesn't tell you is whether the QMS is actually alive day-to-day, maintained, audited internally, enforced on projects, or whether it exists mainly to pass the surveillance audit and then gets ignored until the next one.
The difference shows up in practical ways. In a firm with a functioning QMS, quality isn't something engineers do at the end of a phase to produce documentation. It's woven into how projects run: risk management updated as the design evolves, non-conformances tracked, equipment calibration maintained, design reviews conducted with real scrutiny.
What to ask
Do you have a dedicated quality team: people whose primary role is maintaining and applying the QMS, not engineering?
How does your quality team interact with project engineers day-to-day? (You want to hear: reviews, audits, support, not just sign-off at milestones.)
Does your ISO 13485 certification cover design and development specifically, or only manufacturing?
When was your last internal audit? Your last surveillance audit?
How do you handle non-conformances during a project?
A quality team that runs blitz audits during active projects. Engineers who know how to write a non-conformance report without being told. Equipment calibration logs that are current. Risk files updated at each design review, not assembled retrospectively at the end. They're what ISO 13485 is actually supposed to produce.
Protomed has a dedicated quality team. Their job is to maintain the QMS, support project engineers in applying it correctly, run internal audits, and make sure the system doesn't drift between external surveillance visits. The certification covers design and development, the full development scope, not just manufacturing operations.
3. Do they have in-house prototyping or do they outsource it?
This one matters more than it might seem. Prototyping is not just manufacturing, it's iteration. The number of times you can physically test a concept before your budget runs out is directly tied to how fast and how cheaply a prototype can be produced.
If your engineering partner outsources prototyping to a third party, you're adding lead time, communication overhead, and cost to every iteration cycle. A project that needs eight(y) prototype versions (not unusual) becomes very expensive very quickly.
What to ask
What prototyping capabilities do you have in-house?
What do you outsource, and to whom?
What's your typical turnaround for a functional prototype iteration?
Do you do bench testing in-house, or does that also go to a third party?
-> In-house capabilities worth checking for specifically:
3D printing (including flexible/elastomeric materials if relevant to your device)
CNC machining, laser cutting for metal components
Extrusion or overmolding if the device involves tubing
Basic flow testing, pressure testing, or fatigue rigs
Protomed has in-house prototyping and bench testing. We can go from CAD to a testable prototype in days, for most standard device architectures. For manufacturing processes we don't run ourselves (e.g. laser cutting, industrial extrusion), we work with a stable network of partners we've used on multiple projects.
4. Do they design for your regulatory endpoint or do they design first and think about regulation later?
This is one of the most expensive mistakes in medical device development. An engineering team builds a beautiful device. Then the regulatory consultant gets involved and points out that the chosen materials don't have the right biocompatibility data, or the sterilization method is incompatible with a component, or the device falls into a higher risk class than anticipated because of how a feature was implemented.
Now you're redesigning.
The regulatory endpoint (CE marking under MDR, FDA 510(k) or PMA, or both) should be defined before as early as possible. The choice of target market, device classification, and intended use directly shapes design decisions.
What to ask
At what point in the project do you bring in regulatory considerations?
Do you have internal regulatory expertise, or do you recommend external consultants?
How do you handle biocompatibility strategy, ISO 10993 risk assessment, material selection, testing requirements?
Have you taken a device through a notified body technical file review? What came back?
Red flag: "Quality is one person's part-time responsibility, and that person isn't quality-trained."
In smaller firms, this is common. One engineer "handles quality" alongside a full project load, often without formal quality training. It doesn't mean the certification is fraudulent, but it does mean the QMS is probably reactive rather than preventive, and key quality decisions may lack the expertise they require. Ask who specifically is responsible for quality, what percentage of their time goes to it, and whether they have formal quality training or certification.
Protomed designs with the regulatory endpoint in mind from day one. We have internal knowledge of MDR requirements and maintain active working relationships with regulatory consultants for CE pathway support. For FDA submissions, we work with experienced US regulatory partners.
5. Do they know their limits and do they have the right partners to fill them?
No engineering firm covers everything. The honest ones know exactly where their competence ends. The dangerous ones don't or won't tell you.
Specialization is a feature, not a gap. A firm focused on catheter mechanics is better at catheter mechanics than a generalist. But a focused firm also needs to be honest: clinical trials, advanced material characterization, regulatory submissions, industrial sterilization these are disciplines that require dedicated expertise. A good development partner knows this and has already built relationships with the right specialists.
What you're looking for is a firm that is genuinely expert in their domain and has a reliable, tested network for everything else. Not a firm that does everything superficially, and not one that covers development well but leaves you stranded when you need to move to the next phase.
Phases to map explicitly
Concept and feasibility
Design development and iteration
Biocompatibility and material selection
Verification testing (does the device meet its design specifications?)
Validation testing (does it meet user needs under simulated use conditions?)
Regulatory documentation and technical file preparation
Clinical study support (if required)
For each phase, ask: do you own this, support it, or hand it off? And for the handoffs: to whom, specifically? Have you worked with them before? On how many projects?
Protomed covers concept through design freeze with full regulatory documentation for CE (MDR). We are based at the IHU (Institut Hospitalo-Universitaire) in Strasbourg a clinical research institution that specializes in, among other things, minimally invasive procedure development and clinical trials. That proximity isn't a coincidence. When a project reaches the clinical phase, we have a direct working relationship with the people who run those trials not a cold introduction. For industrialization, we maintain relationships with CDMOs selected for specific manufacturing capabilities, not for convenience.
6. Do they know your type of device AND your clinical domain or just one of them?
Medical devices is an extremely broad category. A wound dressing is a medical device. So is a transcatheter heart valve. So is an MRI machine. So is a glucose test strip.
Between those extremes lies an enormous range of engineering disciplines, materials, mechanics, and clinical environments that have very little overlap. A firm expert in external electronic sensors (wearables, monitoring devices) has a completely different skillset from one that develops implantable mechanical systems for minimally invasive procedures. The physics are different, the materials are different, the failure modes are different, the manufacturing constraints are different.
It goes further. Even within interventional devices, orthopedics and neurology don't share much. Orthopedic implants deal with bone mechanics, load bearing, osseointegration. Neurovascular devices operate in vessels measured in millimeters, with flow dynamics and radiopacity as critical parameters. An engineer who's spent ten years on hip prostheses is not automatically equipped to develop a neurovascular microcatheter. The instincts don't transfer cleanly.
This means you need to check two things, not one: does the firm have experience with your type of device, and do they have experience in your clinical domain?
What to ask
How many projects have you completed in this specific indication?
What are the typical failure modes for this type of device, in your experience? (Listen for specificity — generic answers are a tell.)
What clinical constraints drive the design in this domain — anatomy, procedure workflow, physician ergonomics?
What materials have you qualified for this application, and what drove those choices?
Red flag: adjacent experience presented as equivalent.
"We've done a lot of cardiovascular work" is not the same as neurovascular experience. "We have experience with implants" doesn't mean experience with your specific device architecture. Ask for the specific device types, not the general category. The difference between what they've done and what you need may be larger than it appears.
Protomed's scope is deliberately narrow: cardiovascular (guidewires, catheters, balloon systems, stent delivery), neurovascular (access catheters, coil delivery systems), gastroenterology (endoscopic accessories, biliary devices), and ENT. All minimally invasive. All internal mechanical systems. If your device sits outside this space, we'll tell you that upfront, and we'd rather do that than take a project and figure it out at your expense.
7. Are their engineers the ones you'll actually work with or will the project be handed to someone else once you've signed?
This is one of the most common disappointments in engineering services, and it rarely gets talked about openly.
You have three or four meetings with a senior engineer. The conversation is sharp. They understand the device, they ask good questions, they spot a design risk you hadn't considered. You sign. The project starts. And then, gradually or suddenly, you realize you're now working with someone much more junior, and the person you evaluated is nowhere to be found on your project.
It's not necessarily dishonest. Large firms have business development people whose job is to win projects, and engineers whose job is to execute them. Those are different people. But the gap between the capability you evaluated and the capability actually deployed on your device is real, and it matters.
Even in smaller firms, a senior engineer might be involved in scoping, then largely absent once execution starts, checking in occasionally but not doing the work. You need to know.
What to ask
Who specifically will be the lead engineer on my project, by name?
Can I meet them before we sign anything?
What's their background, how long in medtech, what devices have they personally led?
Who reviews and signs off on technical deliverables?
What is the partner or senior engineer involvement after project kickoff, weekly? On milestones only? Available if needed?
Red flag: you can't get a name.
"Our team has extensive experience" is not an answer to "who will be the lead engineer on my project." If a firm can't or won't name the person who will own technical decisions on your device, that's worth taking seriously. Either they don't know yet (which means the allocation is opportunistic, not planned) or the person doing the selling is not the person doing the work.
At Protomed, the engineers you talk to in scoping are the engineers who run your project. We're a compact team by design. It means we can't take on everything, but it also means there's no version of events where the person you evaluated hands off your device to someone you've never met.
8. What do they do before they give you a plan and do they start by understanding the problem or by scoping the work?
Medical device development takes longer and costs more than most people expect. That's a given. The firms that tell you what you want to hear at the proposal stage are the ones who have difficult conversations six months in, when the schedule has slipped and the original assumptions turned out to be wrong.
But there's a more fundamental question underneath the timeline discussion: how does a firm actually start a project?
A lot of engineering firms will scope the work you described and give you a plan for that. If you say "I need to develop a catheter with this mechanism," they'll plan a catheter development. The problem is that the thing you described may not be the right solution to the problem you're actually trying to solve. Maybe a different mechanism gets you there faster. Maybe a different access route changes the device architecture entirely. Maybe there's a simpler approach that emerged two years ago in an adjacent indication and nobody told you about it.
Before a plan, there should be an exploration. Not unlimited, not unstructured but deliberate. A good engineering partner doesn't rush to scope the solution before mapping the problem.
What to ask
How do you start a project, what's the process before you produce a development plan?
Have you ever told a client that their initial concept wasn't the right approach? What happened?
How do you handle a situation where the scoping phase reveals the project is more complex than anticipated?
What's in scope in the initial phase is it just a technical feasibility, or does it include a review of alternatives?
Before any budget or planning, we run what we call an innovation sprint. A structured exploration phase combining Design Thinking and TRIZ methodology. The goal is not to plan the project you came in with. It's to find the Most Viable Path (MVP and yes, we mean path, not product). That means mapping the full space of possible solutions, identifying constraints, challenging assumptions, and arriving at a development approach that is technically sound and commercially realistic before a single line of the project plan is written.
A sprint typically takes 3 days. It can change the direction of a project significantly. We think it's the most important thing we do.
Rough timeline reference (not guarantees)
Innovation sprint to defined MVP: 3 days
Concept to first functional prototype: 3–6 months
Functional prototype to bench validation: 6–18 months
Full development cycle to regulatory submission: 18–48 months
These ranges are wide because the variables are real. Device complexity, regulatory class, clinical evidence requirements, and the state of the initial concept all have large effects on timeline. Be skeptical of anyone quoting tight timelines without knowing your device, and be equally skeptical of anyone who skips the exploration phase to get to the plan faster.
Summary checklist
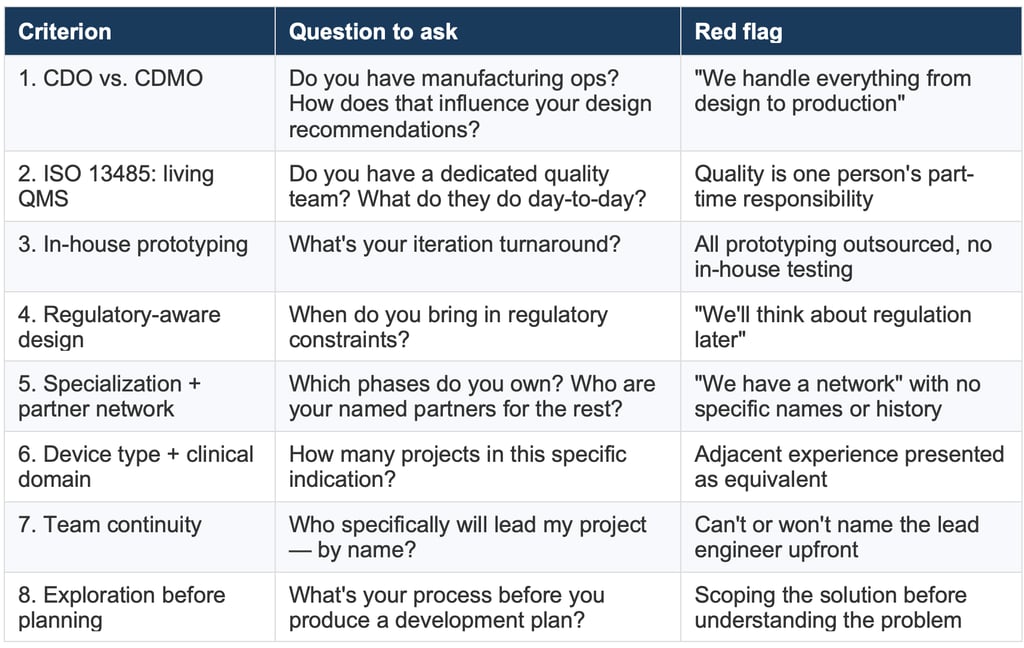
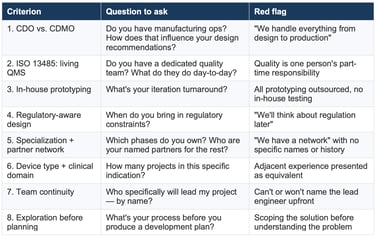
FaQ
1. What is the difference between a CDO and a CDMO for medical devices?
A CDO (Contract Development Organization) handles design and development. Their business is engineering, concept, prototyping, verification, validation, regulatory documentation. They have no manufacturing operations and no commercial incentive to steer designs toward particular production processes. A CDMO (Contract Development and Manufacturing Organization) is primarily an industrial operation. They offer development services, but their core business is manufacturing at scale. The practical implication: a CDO is structurally agnostic about how your device gets made. A CDMO has a factory that needs work, which can influence technical recommendations, not always consciously, but structurally.
2. Is ISO 13485 certification mandatory for a medical device engineering partner?
Not legally mandatory in most jurisdictions but practically essential for any serious project. ISO 13485 requires a Quality Management System covering design controls, risk management, document control, and corrective actions. Without it, your design history file may not satisfy notified body or FDA requirements. More importantly: ask whether the QMS is actively applied day-to-day, or whether it exists mainly to pass the annual surveillance audit. The difference between the two is significant, and it won't be obvious from the certificate alone.
3. How long does medical device development typically take?
From concept to regulatory submission: 36 to 60 months is a realistic range for Class IIa/IIb devices under MDR. The spread is wide because device complexity, clinical evidence requirements, and the state of the initial concept all have large effects on timeline. Simple product improvements with strong predicate devices can move faster. Novel devices with limited clinical literature take longer.
4. What should be in a design history file (DHF)?
A DHF is the complete record of the design and development process, required under ISO 13485 and MDR. It should contain: user needs, design inputs, design outputs, design reviews, verification records (did the device meet its design specifications?), validation records (did the device meet user needs?), and design transfer records. Each element should be linked and traceable. A DHF is not a folder of documents it's a structured, traceable record of design decisions and their rationale.
5. How much does medical device development actually cost?
Medical device development is expensive. Not "startup expensive" — genuinely, structurally expensive.
A useful reference point: a medtech startup burns through roughly €10,000 per working day across its lifetime, when you account for salaries, engineering services, testing, regulatory work, and the inevitable delays. That's not a figure to scare you. It's a figure to help you plan honestly.
Almost no device gets developed without external funding. Some founders can self-finance the early stages — a feasibility study, a first prototype, proof of concept. That's realistic. But by the time you're running bench validation and preparing regulatory documentation, you almost certainly need investors.
The model that actually works is phase-by-phase financing. Each development phase produces concrete deliverables: a validated prototype, a bench test report, a completed risk file. Those results are what you take to your next funding round. You're not asking investors to fund the whole project — you're showing them what you've built, and asking them to fund the next proof point.
This is worth discussing with your engineering partner early. A good partner understands this model and can help you structure the project phases to produce meaningful, investable milestones, not just engineering deliverables. The question isn't only "what does phase 2 cost?" but "what does phase 2 prove, and who will pay for phase 3 once we have it?"



What a real QMS looks like in practice
Red flag: "We have a network of partners" with no names and no history
A partner network isn't a list of companies the firm could theoretically call. It's a set of relationships built through actual project work knowing which CRO handles biliary devices well, which notified body has a reasonable process for neurovascular Class IIb submissions, which CDMO has extrusion capability for PEBA tubing. If they can't be specific, the network may not really exist.
What Protomed calls the "Maze of possible"?


About Protomed
Protomed is an ISO 13485-certified engineering firm based in Strasbourg, France, specializing in minimally invasive medical device development. We work on cardiovascular, neurology, gastroenterology, and ENT devices from concept to design freeze, including full regulatory documentation for CE marking under MDR.
Yann Hoffbeck is Customer Success Manager and CMO at Protomed.
His background is deliberately unusual for a medtech firm. He started as an IT engineer in large banking groups, moved into television journalism, then founded a corporate production company in 2007 that grew into a content-focused communications agency, eventually acquired by a national group, where he became Creative Director. For over a decade, he specialized in communication strategy for technical and scientific environments, working extensively with biomedical laboratories and healthcare organizations.
In a sector that often communicates badly about things it does well, that experience turned out to matter. He left agency life to join Protomed as a consultant and stayed, because the problem of explaining complex engineering work to the people who need it is harder and more interesting than it looks.
About the autor


© 2026. All rights reserved.
Site map
Contract medical device development in cardiovascular and minimally invasive surgery.

