

ISO 13485 : Intégrer les cadres réglementaires dès le premier jour dans le développement de dispositifs médicaux.
Découvrez pourquoi l'intégration précoce de l'ISO 13485 est une approche stratégique pour la R&D MedTech. Assurez la conformité CE marking et FDA pour vos dispositifs chirurgicaux mini-invasifs dès le départ.
Camille S. Quality Manager Regulatory Compliance Engineer
2/17/20266 min temps de lecture
Le développement de dispositifs médicaux, en particulier dans le domaine spécialisé de la chirurgie mini-invasive (MIS), nécessite un alignement précis entre l'innovation en ingénierie et la sécurité clinique. Pour les ingénieurs R&D, les fondateurs de startups et les cliniciens, la transition d'un prototype fonctionnel vers un dispositif médical commercialisable et certifié implique de naviguer dans un environnement réglementaire complexe. Au cœur de ce parcours se trouve l'ISO 13485, la norme internationale pour les Systèmes de Management de la Qualité (QMS) des dispositifs médicaux. Établir ce cadre dès les premières étapes du développement est une décision stratégique qui soutient à la fois l'excellence technique et la scalabilité commerciale.
Comprendre la norme ISO 13485
L'ISO 13485 est une norme QMS autonome, dérivée de la série ISO 9000 mais adaptée spécifiquement à l'industrie des dispositifs médicaux. Elle fournit un cadre complet permettant aux organisations de démontrer leur capacité à fournir des dispositifs médicaux et des services associés qui répondent de manière constante aux exigences des clients et aux exigences réglementaires applicables. Contrairement aux normes qualité générales, l'ISO 13485 met l'accent sur la gestion des risques, les contrôles de conception et les environnements de production, des éléments essentiels dans le développement d'instrumentation chirurgicale.
La norme s'applique à toutes les étapes du cycle de vie, notamment la conception et le développement, la production, le stockage et la distribution, l'installation et la maintenance. Pour une startup ou un département R&D, adopter l'ISO 13485 signifie mettre en place une approche systématique de la documentation et du contrôle des processus. Cela garantit que chaque décision prise lors du développement d'un dispositif est traçable, reproductible et axée sur la sécurité du patient.
Distinguer l'ISO 13485 du Medical Device Regulation (MDR)
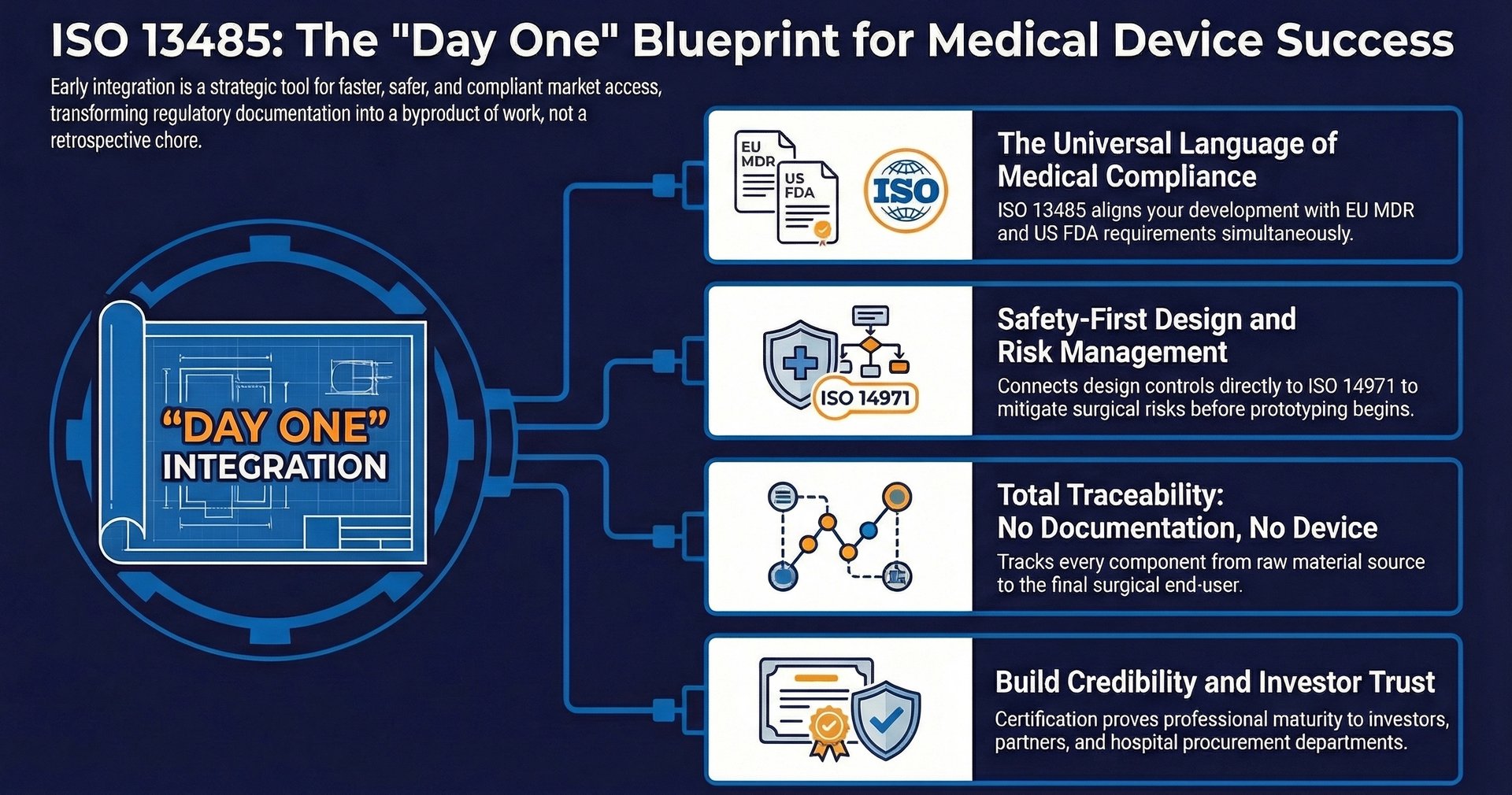
Un point de discussion fréquent dans le secteur médical européen est la relation entre l'ISO 13485 et le Medical Device Regulation européen (MDR 2017/745). Bien qu'ils soient distincts, ils fonctionnent de manière complémentaire. Le MDR est une exigence légale : un règlement qui doit être respecté pour mettre un dispositif sur le marché européen. Depuis son harmonisation avec le MDR 2017/745, la conformité à l'ISO 13485:2016 permet de satisfaire une grande partie des exigences QMS du règlement.
Choisir l'ISO 13485 comme fondation de votre système qualité est la voie la plus reconnue vers la conformité. Tandis que le MDR précise ce qui doit être accompli pour mettre un dispositif médical sur le marché (comme l'évaluation clinique et la surveillance post-commercialisation), l'ISO 13485 décrit comment gérer les processus internes pour atteindre cet objectif. L'ISO 13485 est la norme mondiale pour les dispositifs médicaux, et de nombreuses réglementations locales sont basées sur ses dispositions ou les complètent, comme le Quality System Regulation (QSR) de la FDA aux États-Unis.
Le rôle de l'ISO 13485 dans le CE marking et la FDA Clearance
Pour qu'un dispositif médical atteigne le milieu clinique, il doit être évalué par des organismes réglementaires : les Notified Bodies pour le CE marking en Europe, ou la FDA pour les voies 510(k) ou PMA aux États-Unis. L'ISO 13485 sert de langage universel entre le fabricant et ces autorités.
Dans l'Union européenne, la conformité à l'ISO 13485 crée une présomption de conformité aux exigences QMS du MDR. De même, la FDA a récemment aligné sa réglementation 21 CFR Part 820 plus étroitement avec l'ISO 13485 à travers le Quality System Regulation Amendment (QSRA).
En mettant en œuvre l'ISO 13485 dès le début, une entreprise constitue la Technical Documentation et le Design History File (DHF) requis pour ces soumissions de manière naturelle et progressive. Cet alignement facilite des audits plus fluides et réduit le temps nécessaire à l'examen réglementaire.
Mise en œuvre pratique : QMS, normes et reporting — Piliers essentiels pour un accès rapide et sécurisé au marché
En pratique, la mise en œuvre de l'ISO 13485 dès le départ implique d'établir plusieurs piliers fondamentaux au sein de l'organisation. Ces piliers fournissent la structure nécessaire aux projets à forts enjeux comme les outils chirurgicaux mini-invasifs. Bien qu'ils couvrent des domaines tels que la formation du personnel et les engagements de la direction, nous mettons ici en avant ceux qui sont pertinents pour un développement réussi.
Contrôles de conception et gestion des risques
La norme exige un processus de conception et de développement rigoureux. Cela comprend des design inputs définis (ce que le dispositif doit faire), des design outputs (une conception et ses spécifications), ainsi que des activités de vérification et de validation. La vérification garantit que le dispositif est construit correctement, tandis que la validation s'assure que le bon dispositif a été conçu pour les besoins de l'utilisateur. On peut notamment souligner le lien avec l'ISO 14971 sur la gestion des risques, qui permet l'identification et la mitigation des risques dès le début du projet, en favorisant l'identification la plus précoce possible des modes de défaillance potentiels dans une procédure chirurgicale, dès le stade des design inputs.
Documentation contrôlée et traçabilité
L'ISO 13485 souligne que « si ce n'est pas documenté, cela n'a pas eu lieu ». Cela implique de maintenir un Device Master Record (DMR) qui contient les instructions pour la fabrication et les tests du dispositif. Il est également question de maintenir le dossier du dispositif médical, qui permettra de retracer toute la vie du dispositif, depuis sa dernière amélioration commercialisée jusqu'à l'origine de l'idée initiale.
La traçabilité n'est pas seulement une question de conception, mais aussi de routine : elle est tout aussi vitale au quotidien ; pour un dispositif MIS, cela signifie être capable de retracer chaque composant jusqu'à sa source de matière première et jusqu'à l'utilisateur final.
Corrective Actions and Preventive Actions (CAPA)
Le QMS comprend une boucle de rétroaction connue sous le nom de CAPA. Ce processus permet à l'équipe d'éliminer toute non-conformité, d'en investiguer les causes profondes, qu'elles soient identifiées lors des tests ou via les retours des cliniciens, et de mettre en place des changements pour éviter toute récurrence. Cela crée une culture d'amélioration continue qui renforce la fiabilité du dispositif.
Les avantages de l'intégration réglementaire dès les premières étapes
Intégrer le cadre réglementaire dès le « Jour 1 » est souvent perçu sous l'angle de l'efficacité. Lorsqu'une équipe de développement opère dans un environnement conforme à l'ISO 13485 dès le début, la documentation requise pour une soumission réglementaire devient un sous-produit naturel du travail, plutôt qu'une tâche séparée et rétrospective.
L'intégration précoce permet d'identifier les « Vertical Standards » applicables, des normes spécifiques à certains types de dispositifs, comme l'ISO 60601 pour les équipements médicaux électriques ou l'ISO 10555-1 pour les cathéters stériles à usage unique. Connaître ces exigences dès la phase de conception initiale évite des choix de design qui pourraient s'avérer non conformes par la suite. Cette approche proactive garantit que le chemin du laboratoire à la salle d'opération est prévisible et caractérisé par une progression constante.
Premières étapes pour établir un cadre ISO 13485
Démarrer le parcours vers la conformité ISO 13485 est un processus gérable lorsqu'il est décomposé en phases logiques. Pour de nombreuses startups et équipes R&D, la voie la plus efficace combine engagement interne et expertise externe.
Gap Analysis : Évaluer les processus existants par rapport aux exigences de la norme afin d'identifier ce qui est déjà en place et ce qui doit être développé.
Définir le périmètre : Préciser clairement les types de dispositifs et les activités que le QMS couvrira. Pour une entreprise axée sur la MIS, cela inclurait la conception et potentiellement le prototypage en petites séries.
Établir la politique et les objectifs qualité : Aligner les objectifs de l'organisation avec un engagement envers la qualité et la conformité réglementaire.
Process Mapping : Documenter les processus clés de l'organisation, depuis la capture des exigences des cliniciens jusqu'à la sélection et la gestion des fournisseurs.
Formation : S'assurer que les équipes d'ingénierie et cliniques comprennent leur rôle au sein du QMS. La conformité est une responsabilité collective.
Bâtir la crédibilité grâce à la rigueur technique
Pour les fondateurs et les cliniciens, l'objectif ultime est d'amener sur le marché une solution qui améliore les résultats pour les patients. Un cadre certifié ISO 13485 est un puissant indicateur de maturité professionnelle. Il démontre aux investisseurs, partenaires et services d'approvisionnement hospitaliers que le dispositif a été développé dans un environnement contrôlé et de haute qualité. Dans le domaine de la chirurgie, où la précision est primordiale, cette rigueur technique est le fondement de la confiance.
En traitant le cadre réglementaire comme un outil pour une meilleure ingénierie plutôt que comme un obstacle bureaucratique, les équipes peuvent se concentrer sur ce qu'elles font le mieux : innover pour l'avenir des soins de santé.


© 2026. Tous droits reservées.
Plan du site
Mentions légales
Développement de dispositifs médicaux sous contrat pour la chirurgie cardiovasculaire et mini-invasive

